

In the ever-expanding catalog of differences between humans and our primate cousins, cancer risk rarely tops the list. Yet humans, despite sharing over 98% of our DNA with chimpanzees, are significantly more prone to developing solid tumors. A recent study from UC Davis, published in Nature Communications1, adds an unexpected twist to the story of human susceptibility. It traces our increased cancer vulnerability to a single amino acid change in a critical immune protein: Fas Ligand (FasL).
FasL is central to a process called apoptosis, a kind of cellular self-destruct button. When immune cells such as T lymphocytes or CAR-T cells detect a malignant cell, they use FasL to trigger its death. The process is fast, precise, and normally effective. But in humans, a tiny evolutionary change appears to have given tumors a loophole.
The Plasmin Problem
The study zeroes in on a mutation that distinguishes the human version of FasL from that of other primates. Where chimpanzees have a proline at position 153 in the protein's amino acid sequence, humans carry a serine.
"A single Pro153-to-Ser153 substitution makes human FasL uniquely vulnerable to cleavage by plasmin," the authors report.
Plasmin is a protease, an enzyme that breaks down proteins, and it’s often abundant in the environment around solid tumors. This makes human FasL a sitting duck. Once cleaved by plasmin, FasL loses its killing edge. The immune cell is still there, ready for action, but one of its sharpest weapons has been disarmed.
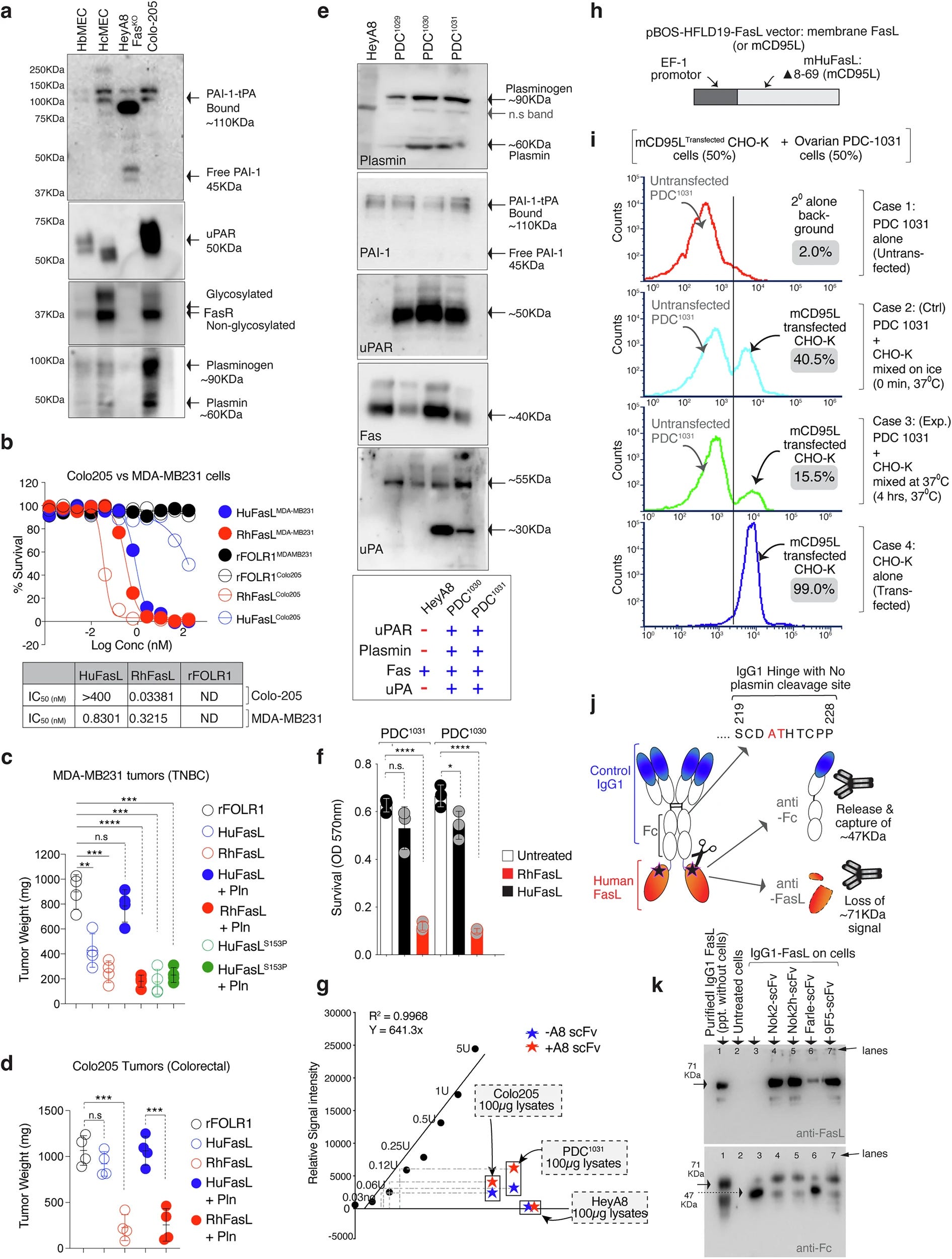
This vulnerability doesn’t show up in chimpanzees, rhesus macaques, or mice, which have a structurally more robust FasL. In these species, FasL resists plasmin’s attacks. That resistance may help explain why non-human primates tend to develop fewer aggressive solid tumors.
A Cost of Being Human?
The researchers hypothesize that the serine substitution in human FasL might not be a random accident. Evolution often involves tradeoffs. The mutation could have played a role in shaping aspects of human biology beyond immunity, perhaps contributing to the development of our large brains or finely tuned immune responses. But when it comes to cancer defense, the change is clearly a liability.
FasL’s susceptibility to cleavage effectively allows tumors to turn off a key death signal. This helps explain why T cell-based immunotherapies, especially CAR-T therapies that work well in blood cancers, often struggle in solid tumors. The immune system wants to fight, but the tumor has found a molecular escape route.
Blocking the Break
What if that loophole could be closed? The researchers explored this question too. In lab experiments and mouse models, they tested two strategies: inhibiting plasmin itself and protecting FasL with antibody fragments that block cleavage.
Both approaches worked. Plasmin inhibitors like aprotinin restored FasL’s tumor-killing capacity. So did two antibodies, Nok2h and 9F5, which bind near the cleavage site and prevent the enzyme from gaining access. In one experiment, treating tumors with human FasL plus a plasmin-blocking antibody led to a noticeable return of T cell-mediated tumor destruction.
"These antibodies did not interfere with FasL binding to its receptor but successfully shielded it from enzymatic attack," the authors wrote.
The finding suggests that part of the immunotherapy puzzle in solid tumors might be solved by fine-tuning the FasL pathway.
An Ancient Legacy in Modern Medicine
This research does more than open the door to better cancer treatments. It reminds us that the human genome is a palimpsest, written over millions of years and still bearing the marks of its evolutionary past. Some of those marks, like the Pro153-to-Ser153 mutation, once carried benefits or were at least neutral. Today, in the context of cancer biology, they pose new challenges.
"This study exemplifies how evolutionary history continues to shape human disease," noted the team from UC Davis.
Looking Forward
The next steps will likely involve developing therapies that combine T cell-based approaches with FasL stabilization. The antibodies Nok2h and 9F5, already effective in animal models, could be engineered into bispecific formats or fused with FasL itself for enhanced potency.
There’s also a broader question: how many other seemingly minor human-specific mutations have unexpected medical consequences? As comparative genomics continues to map the subtle differences between species, the evolutionary roots of disease may become a rich field for discovery.
Related Research:
Chen, D. S., & Mellman, I. (2017). Elements of cancer immunity and the cancer–immune set point. Nature, 541(7637), 321–330. https://doi.org/10.1038/nature21349
Hasin-Brumshtein, Y., Lancet, D., & Olender, T. (2009). Human olfaction: from genes to behavior. Human Genomics, 3(1), 67. https://doi.org/10.1186/1479-7364-3-1-67
Nagata, S. (1997). Apoptosis by death factor. Cell, 88(3), 355–365. https://doi.org/10.1016/S0092-8674(00)81874-7
Legembre, P., Schickel, R., & Barnhart, B. C. (2006). Regulation of FasL and Fas by posttranslational modifications. Cell Death & Differentiation, 13(8), 1215–1223. https://doi.org/10.1038/sj.cdd.4401940
Wamba, B. E. N., Mondal, T., Freenor, F., V., Shaheed, M., Pang, O., Bedinger, D., Legembre, P., Devel, L., Bhatnagar, S., Leiserowitz, G. S., & Tushir-Singh, J. (2025). Evolutionary regulation of human Fas ligand (CD95L) by plasmin in solid cancer immunotherapy. Nature Communications, 16(1), 5748. https://doi.org/10.1038/s41467-025-60990-0






