
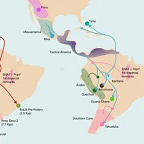
When researchers tried to model the ancestry of present-day Indigenous South Americans using two source populations, the statistics said no. Not just inadequately — flatly. A two-source model was rejected at P = 0. A minimum of three genetically distinct ancestral streams, from three separate dispersal events, was required to account for what’s in living genomes.
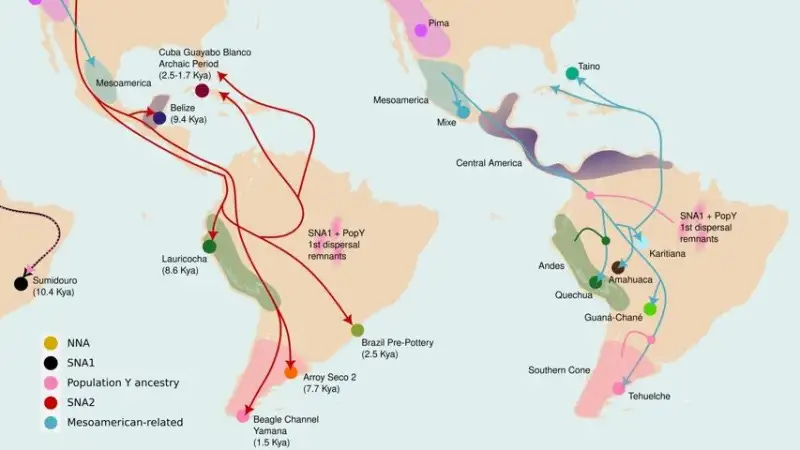
That’s the core finding of a new Nature1 paper from the Indigenous American Genomic Diversity Project, and it means the standard account of how people came to populate South America is missing a chapter.
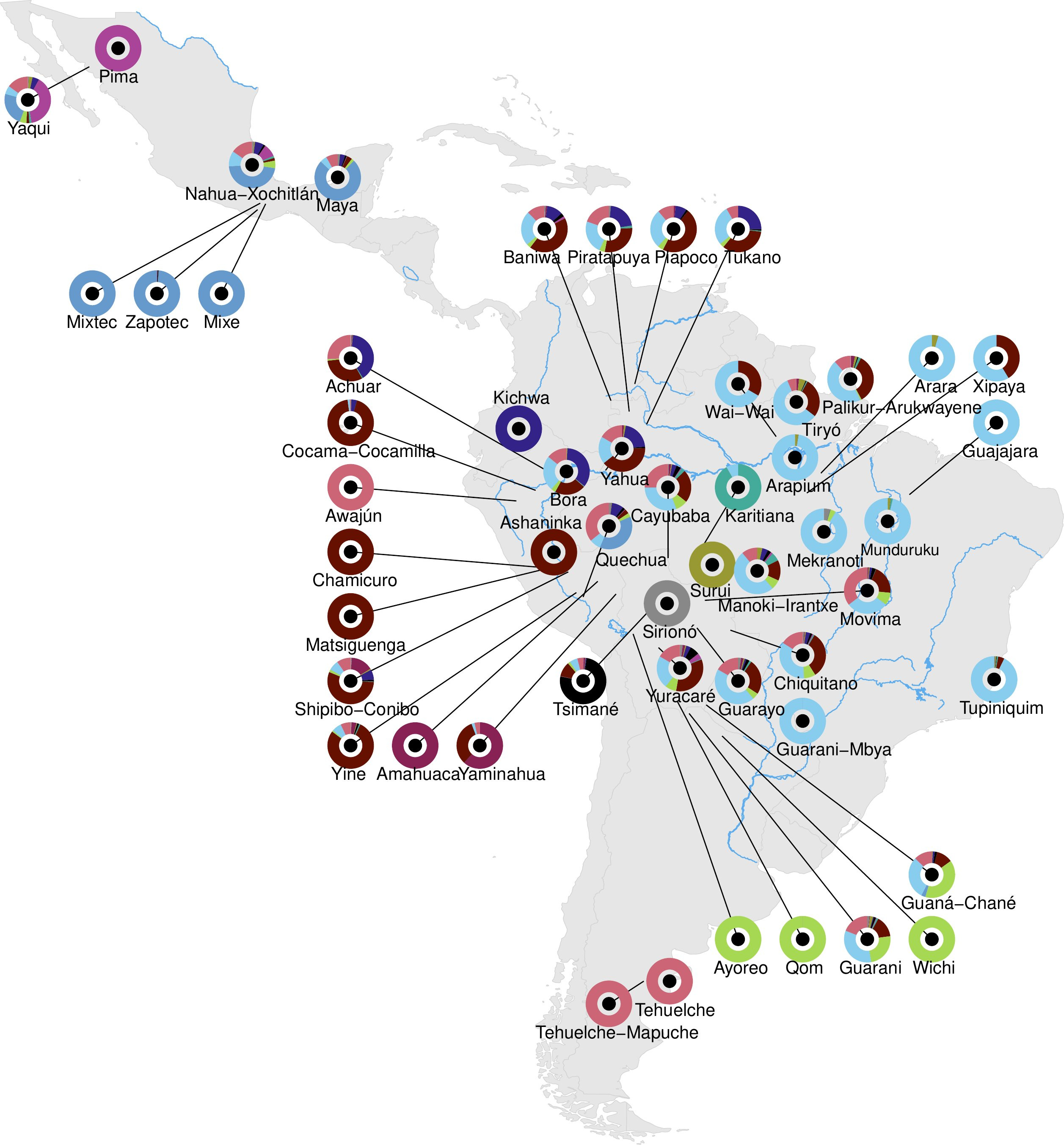
The study, led by Marcos Araújo Castro e Silva, Tábita Hünemeier, David Comas, and colleagues at the Institute of Evolutionary Biology in Barcelona and the University of São Paulo, presents 128 new high-coverage whole-genome sequences from Indigenous individuals across eight Latin American countries — Argentina, Bolivia, Brazil, Colombia, Ecuador, Mexico, Paraguay, and Peru — spanning 45 populations and 28 linguistic families. Combined with existing published data, the final dataset covers 199 individuals from 53 populations and 31 linguistic families. Average sequencing depth was around 44 times, sufficient for high-confidence variant calling. Ancient DNA from the Allen Ancient DNA Resource and previously published Brazilian sambaqui genomes was incorporated to anchor population history across time.
The total variant count: 12.49 million single-nucleotide variants, of which 1.43 million — about 11.4% — weren’t recorded in any major public database, including gnomAD, the 1000 Genomes Project, or dbSNP. That’s roughly 11,100 novel variants per individual. The scale of what was missing from global genomic resources isn’t a minor data gap.
Part of the reason for that gap is methodological. Genomic studies of Indigenous Americans have often relied on a small number of well-characterized groups as stand-ins for broader diversity. The Karitiana and Suruí from the Brazilian Amazon have been especially common proxies for lowland South American populations. This study’s analyses show they’re poor ones. Both groups carry unusually high inbreeding coefficients — more long identical-by-descent segments than geographically nearby groups — and cluster in ways that separate them even from populations living close by. Using them as proxies for Amazonian diversity, the authors argue, systematically distorts inference. Their genetic distinctiveness isn’t ancient; it’s recent, driven by population collapse after European contact.








